Pharmacophore an International Research Journal
CURRENT REVIEW ON IRS-1, JNK, NF-ΚB & m-TOR PATHWAYS IN INSULIN RESISTANCE
Nikita Saraswat1*, Pranay Wal2, Rashmi Saxena Pal1, Ankita Wal2, Yogendra Pal1, Deepa Maurya3
|
|
Abstract
Keywords: Diabetes mellitus, islet amyloid, insulin resistance, insulin action, IRS-1, beta-cell dysfunction, oxidative stress, JNK and IKKβ, mitochondria dysfunction Introduction
Diabetes is most frequently known as diabetes mellitus, which is a category of metabolic disorder and is one of the parts of ordinary systemic disease in the world [1-3]. Diabetes along with hypertension is one of the most progressing diseases, which is majorly threatening public health as well as the socioeconomic. The mortality and morbidity rate of diabetes is increasing the passage of time [4]. According to the international diabetes federation 2017 report, 425 million of the worldwide population are diagnosed with diabetes and is supposed to elevate by 48% in the year 2045. Diabetes is a long-term disorder of the endocrine system in which the protein, fat, carbohydrates, water and electrolytes metabolism changes extensively because of disbalance insulin production [5]. Diabetes is a condition in which blood glucose level increases after meal malfunction [6, 7]. Deficiency of insulin leads to diabetes, 2.8% world population is being reported to be affected by this disease and the prevalence rate may rise by 2025 to the extent of 5.4% [6, 8]. Diabetes needs to be early diagnosis followed by treatment and a restricted lifestyle. Diabetes is one of the leading causes of death in the 21st century [9]. Diabetes is determined to be a major searching health issue in the 21st century [10-12]. Diabetes has dependably been chronicled as a metabolic disorder signalized by hyperglycemia that progresses as a result of deficiencies in insulin secretion, insulin action or both of them. Diabetes is signalized by immedicable hyperglycemia with discompose of protein, fat and carbohydrate metabolism and is connected with most ordinary long-term diabetes associated with damage and dysfunction and failure of different organs, particularly the eyes, blood vessels, hearts, nerve, and kidney [13-15]. Diabetes is greatly explained as a disorder characterized by unprofessional fasting or post nutrition hyperglycemia, caused by complete or respective insulin defect and its metabolic results, which involve distressed metabolism of protein, fat, and carbohydrate. The worldwide occurrences of diabetes s approximated to increases from 4% in the 1995 year to 5.4% by the 2025 year [16, 17]. Classification of diabetes mellitus: Diabetes mellitus can be classified into three categories: [18]
The American diabetes association has classified the population into four groups such as type 1 diabetes mellitus, type 2 diabetes mellitus, gestational and diabetes associated with a specific condition [19, 20]. Type 1 diabetes is also called insulin-dependent diabetes and 95% of diabetes which is non-insulin dependent or Type 2 diabetes occurs in those who have obesity and insulin resistance correlate this form of diabetes [21-27]. Type 1 DM-diabetes Mellitus: Type 1 diabetes mellitus (T1DM) earlier known as insulin-dependent diabetes mellitus or juvenile diabetes, which is most commonly results from autoimmune destruction of insulin-secreting beta cells in the pancreas [21-27]. Diabetes is defined as insulin-dependent diabetes mellitus (IDDM), the onset of insulin-dependent diabetes mellitus occurs with insulinogenic and to sustain life one has to be dependent on insulin injection. This type of diabetes can also term as juvenile diabetes [28]. Type 2 DM-diabetes Mellitus: Type 2 diabetes mellitus earlier known as non-insulin diabetes mellitus or adult-onset diabetes mellitus, is an adult metabolic disorder that is characterized by peripheral tissues of insulin resistance and pancreatic beta-cell function [21-27]. Type 2 diabetes is also called non-insulin dependent diabetes mellitus (NIDDM), non-insulin dependent diabetes mellitus patient needs to get symptomatic relief from fasting hyperglycemia patients with non-insulin dependent diabetes mellitus are prone to ketonuria. Non-insulin dependent diabetes mellitus patient is not dependent on insulin. Although they require insulin for prevention from ketonuria [28]. GDM-Gestational diabetes mellitus: Gestational diabetes mellitus is the third type of diabetes that is found when the release of high blood glucose levels affects a pregnant woman and usually disappears after giving birth [21]. Diabetes: stages and types of etiology are shown in (table 1) [29].
Table 1. Diabetes of glycemia, stages, and types of etiology [29].
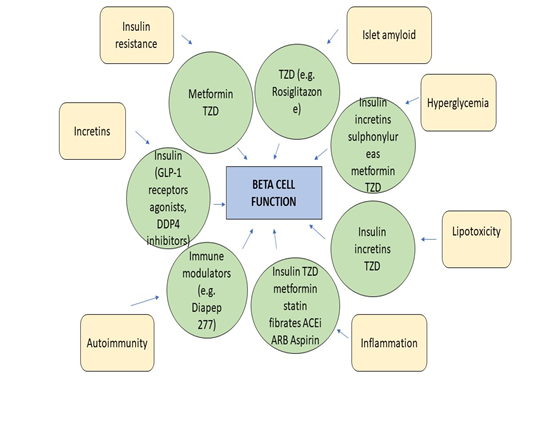
Diabetes stages and types of etiology. *The patient can reverse to normal glycemia condition without any continuous therapy. **The patient may need insulin to stain life. Factors Related to Beta-Cell Dysfunction Various factors are hyperglycemia or glucotoxicity, lipotoxicity, inflammation and autoimmunity, adipokines, islet amyloid, incretins, and insulin resistance. Factors related to beta-cell dysfunction pharmacologic drugs that affect them in (Figure 1) [30]. Increasing to donate loss of beta-cell function in subjects is known to various factors with diabetes [31]. Figure 1. A diagrammatic representation of the factors related to beta-cell dysfunction pharmacologic drugs that affect them. GLP-1–glucagon-likepeptide-1, TZD-thiazolidinedione, DDP4-dipeptidyl peptidase-4, ARB – Angiotensin receptor blocker, ACEiAngiotensin-converting enzyme inhibitors [30].
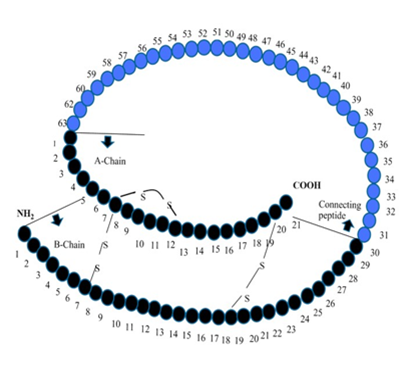
Hyperglycemia or glucotoxicity: Pancreatic beta cells are highly responsive to the concentration of blood glucose variation in blood glucose balance also affect the function of the beta-cell. Insulin secretion has the impact of abnormally elevated blood glucose, the survival of cell sensitive of insulin via a different mechanism, cause hyperglycemia and result in beta cell dysfunction [32]. Lipotoxicity: FFA concentration is correlated with diabetes. Continuous increase. FFA leads to having an unfavorable effect on beta-cell function and deposition of FA metabolize, which toxic at the site of islet cells. Through many experimental observations, it is concluded FFA toxicity is the result of Mani parameters like the nature of chain length unsaturation level and amount of free FFA [33-36]. Autoimmunity and inflammation: Autoimmune place a major role in T1DM (Type 1 diabetes mellitus) due to autoimmune stimulated apoptosis, during disease progressive results in the low secretion of insulin and mass of beta-cell [37]. Several T2DM (Type 2 diabetes mellitus) subject characterizing phenotypes have markers for autoimmunity, which are internally related to the function of beta cells [38-40]. According to reports, autoantibodies are found highly in young as well as leaner individuals [41]. Adipokines: Adipokines are peptides of hormonal nature secreted from adipose tissue that contribute to the deuteration of beta-cell and its function [42]. Islet amyloid: Excessive secretion of IAPP in the pancreatic islets along with amyloid deposits considered to have characterized, which contribute pathological condition for T2DM and destruction of the beta-cell. IAPP is considered a normal beta cell product, which is secreted along with insulin and also gets stored with it in secretory granules. IAPP ‘s actions are not clearly defined through and it is assumed that it inhibits the secretion of insulin, which is based on glucose stimulation. It is also affecting appetite as well as gastric emptying [43, 44]. Incretins: Incretins have the positive effect of cellular action of beta cells by adhering to receptors present on beta cells specifically and elevating intracellular cyclic AMP (CAMP) as well as calcium concentration [45, 46]. It also activates GSIS, which emprises response. Incretins are also enhanced certain genes that are beta cell-specific, including glucokinase, glucose transporters and proinsulin [45-48]. Incretins increase the efficiency of the beta-cell. In the case of T1DM, the secretion of incretin gets impaired and their action gets prohibited, which results in insulin deficiency and causes destruction and dysfunction of beta-cell [45, 49]. Incretin levels are further slowing down due to the de-sensitization of particular receptors or through down-regulation [50, 51]. Insulin Resistance: Insulin was the first hormone discovered by the peptide. Before the level of crystallized insulin from Abel in 1926 and the B-chain N-terminal phenylalanine was identified in 1935 Jensen and Evens, which proves that insulin is a protein and all the hormones are a small molecule. After the spencer's understanding of the amino acid sequence of insulin, it was discovered that insulin was a two-chain connected to a 30-residue b series with the system residue (A7-B7 and A20-B19). One has also been found in the intrachain discharge bond a chain (A6-A11). Insulin biosynthesis and mechanism of release in figure (2), the primary structure of porcine insulin and porcine proinsulin.
Figure 2. A diagrammatic representation of insulin biosynthesis and mechanism of release [52].
Primary premutation of the porcine insulin and insulin. the order of human insulin is similar to that of porcine insulin except by the change of AlaB30 to ThrB30 in human insulin [52]. Due to insulin resistance, the increase in the demand for the high beta-cell function to release insulin leads to cell dysfunction and the chances of failure. The exact mechanism of resistance is not clearly defined but it is postulated or different theories that beta-cell exhaustion may lead to hypersecretion of insulin [53]. Type 2 diabetes and insulin resistance are associated with each other however, records indicate that changes in insulin resistance are also increasing type 1 diabetes [54]. The mechanism of diabetes resistance is not understood but, it is set to be correlated with oxidative stress, environmental, genetic, habitual, inflammation. There is an intensive need for novel approaches to minimize insulin resistance in diabetes. Insulin sensitivity starts to fall due to free fatty acids in the case of type 2 diabetes. The mechanism is not clearly understood but it is said that the higher concentration of fatty acids metabolites activates a cascade of serine kinase which intern alters the signaling process. Resistance is also affected by the dysfunction of several proteins and molecules involved in signaling. Instead of all these postulates, there is a huge need for additional researches to completely define the mechanism of insulin resistance [55-57]. Materials and Methods: A literature search was made on database like PubMed and Medline by using keyword "Diabetes mellitus", "hyperglycemia", "pancreatic beta cells", "glucotoxicity", "lipotoxicity", "adipokines", "incretin", "autoimmunity", "islet amyloid", "insulin resistance", "insulin action", "IRS-1", "PI3-kinase", "beta-cell dysfunction", "oxidative stress", "inflammation", "physical inactivity", "dyslipidemia", "JNK and IKKβ", "mitochondria dysfunction". A combination of research and review paper was found and to get the most suitable article, non-relevant data were excluded. Selection of data has been done by studying combination of research and review papers from different databases like PubMed, Core, Science3open, Directory of open access journals, EMBASE, Europe PMC, FSTA-Food Science and Technology, Nutrition, Google Scholar, Hub Med, Merck Index, MedlinePlus, Indian citation index, ScienceOpen, PubMed, Scopus, Semantic Scholar, WorldWideScience, Shodhganga, Science Direct from year 1985- 2018 by using search keywords like "Diabetes mellitus", "hyperglycemia", "pancreatic beta cells", "glucotoxicity", "lipotoxicity", "adipokines", "incretin", "autoimmunity", "islet amyloid", "insulin resistance", "insulin action", "IRS-1", "PI3-kinase" "beta-cell dysfunction", "oxidative stress", "inflammation", "physical inactivity", "dyslipidemia", "JNK and IKKβ", "mitochondria dysfunction". Inclusion and exclusion of databases 500 publications were searched using keywords “Diabetes mellitus”, “hyperglycemia”, “pancreatic beta cells”, “glucotoxicity”, “lipotoxicity”, “adipokines”, “incretin”, “autoimmunity”, “islet amyloid”, “insulin resistance”, “insulin action”, “IRS-1”, “PI3-kinase”,beta-cell dysfunction, oxidative stress, inflammation, physical inactivity, dyslipidemia, JNK and IKKβ, mitochondria dysfunction in databases PubMed, Core, Science3open, Directory of open access journals, EMBASE, Europe PMC, FSTA-Food Science and Technology, Nutrition, Google Scholar, HubMed, Merck Index, MedlinePlus, Indian citation index, ScienceOpen, PubMed, Scopus, Semantic Scholar, WorldWideScience, Shodhganga, Science Direct from year 1985- 2019. Inclusion 107 References were included because of the following reasons:
Exclusion Rest papers were excluded because of the following reasons:
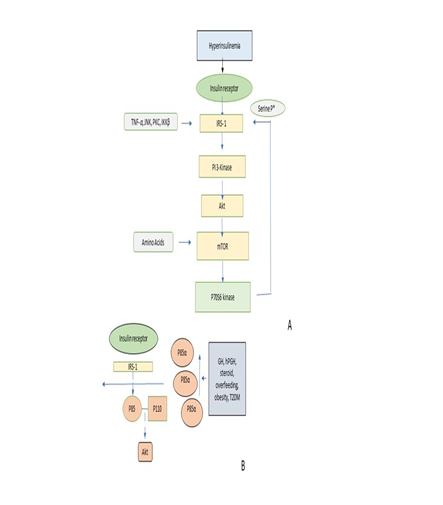
Mechanism Involved in Inducing Insulin Resistance: Insulin resistance is a direct effect of obesity-related exposure of tissues to analyze dietary nutrients, following in the metabolic toxic of collection by-products that are a generally held nation. But more recent work is indicated that factors can also be important are including intercommunication organ networks that are moderated by peptide hormone and molecules inflammatory (cytokines) [58]. It is a molecular mechanism of insulin resistance to survive incompletely understood. Though insulin resistance has appeared as a problem of broad health care, obesity, diabetes, hypertension, and cardiovascular disease [59, 60]. In the mechanism of insulin resistance in diabetes [61], reduced insulin action increased hepatic glucose production and identify insulin secretory. Excepts for beta-cell failure [62], type 2 diabetes mellitus of evolution to the great pathophysiology incident presenting in the target tissue of resistance to insulin [63]. Usually, blood glucose levels of decrease insulin by fat tissue and promote glucose uptake mostly into skeletal muscle, by the liver and endogenous glucose manufacturing of insulin. These organs do not currently respond to insulin, in states of insulin resistance, thereby causing a reactive increase and hyperglycemia in insulin secretion by the pancreatic beta-cell [64]. Analytically, the term indirect insulin resistance that the concentration of insulin is required to be higher or lower to maintain normoglycemia. This violent circle lastly shows disturbance of the balance breakable between beta-cell function and insulin resistance of peripheral which lastly results in the analytical expression of type 2 diabetes mellitus [T2DM] [65]. Anatomically, the production of insulin is released by the pancreatic beta cells of postprandially in sequence to preserve euglycemia. Uptake of glucose is promoted by insulin and insulin facilities transport of glucose into fat and skeletal muscle by activating transporter 4 (GLUT4) from the plasma membrane to the cytosol [66, 67]. Almost, if not completely, of the metabolism effects and antiapoptotic effects of insulin are moderated by the pathway of signaling including IRS proteins, phosphatidylinositol 3-kinase (PI 3-kinase) activation, phosphorylation, mTOR (molecular target of rapamycin), Akt (also called protein kinase B or PKB) and p7056 kinase [68-71]. PI 3-kinase of activation, Akt and uncommon protein kinase (PKC) across the phosphoinositide depend on protein kinase (PDK) [72] materialize to be disapproving in the mechanism and glucose transport [73]. The maintained normoglycemia is required for insulin with normal concentration, which in the clinical term is insulin resistance. Insulin is physiologically related by pancreatic beta-cell postprandially in sequence to maintain euglycemia. At any time, S6K1 kinase and mTOR of activation obtain serine phosphorylation of IRS-1, with the IRS-1-associated PI 3-kinase activity in a subsequent decline (Figure 3) [74].
Figure 3. A diagrammatic representation of the molecular mechanism of insulin resistance by Serine phosphorylation of IRS -1 protein [73].
Serine phosphorylation of IRS-1: Serine phosphorylation of IRS-1 protein through tyrosine kinase is connected with insulin receptors during the binding of insulin. MYMX IRS-1 motif obtains incorporated by PI 3- kinase having a residue of phosphorylated tyrosine kinase. This triggers PI-3 kinase major to a downstream cascade which takes with stimulation and phosphorylation of mTOR and Akt p70S6. Stimulation of Akt is responsible for the transportation of glucose and stimulation of mTOR and p70S6 kinase is responsible for protein synthesis. A. Due to hyperactivation of mTOR through hyperinsulinemia, amino acids, and Akt, serine phosphorylation of pf IRS-1 through p70S6 takes place which intern subside the vitality of PI-3/IRS1 signaling. Additionally, this phosphorylation process of IRS-1 perhaps is augmented by TNF α and β, IKK β, PKC and JNK. B. The heterodimer p85 – p110 is replaced by monomer p85 α antagonistically away from the binding site of IRS-1 protein, which decreases the regulation of PI 3 – kinase [73]. Initially, it becomes evident that the capacity of IRS protein to attract PI 3-kinase is reduced by serine phosphorylation of IRS proteins. Thereby reducing it is activation [75-81]. And serine phosphorylation of IRS proteins can also be conducted to increase the degradation of protein of IRS-1 [82]. Serine phosphorylation can be obtained in reaction to several intracellular serine kinases (Table 2) [83-94].
Table 2: serine phosphorylation can obtain in reaction to several intracellular serine kinases [81-93].
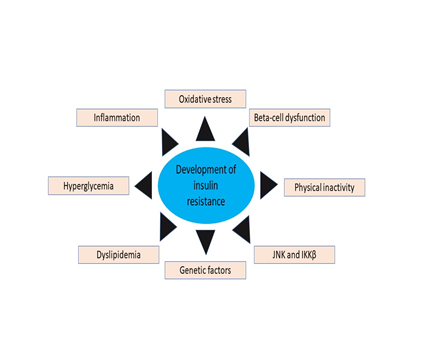
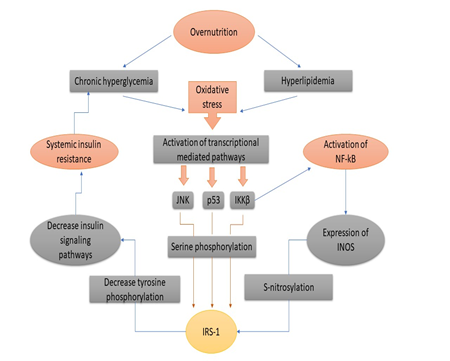
Various Transcriptional and Metabolic Pathways (Figure. 4) [95]: Schematic representation of the development of IR by beta-cell dysfunction, oxidative stress, inflammation, physical inactivity, dyslipidemia, JNK and IKKβ, hyperglycemia, genetic factors (figure. 3) adopted from Rehman K. and Akash M.S.H. [95] Oxidative Stress in Insulin Resistance: Excess nutrition increases the cellular burden of glucose and FFAs, which leads to oxidative stress (Figure 4). Insulin mediation by tissues through JNK and NF-κB pathways reduces glucose and insulin signaling, which ultimately causes the insulin resistance to lay it induces (Figure. 5) [96-98].
Figure 4. A schematic representation of insulin resistance of development which adopted from Rehman K. and Akash M.S.H. [94]
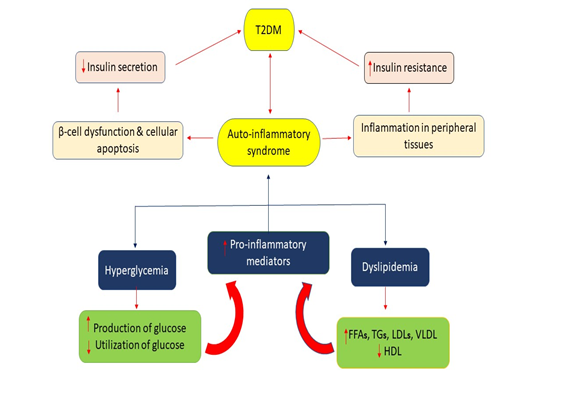
Figure 5. Mechanism of oxidative stress-induced Insulin resistance [95-97]. Mechanism of Oxidative Stress-Induced Insulin Resistance [Figure 5]: Hyperglycemia, due to high nutrition and exposure to hyperlipidemia for long periods, causes oxidative stress due to the activation of reactive oxygen species. Once the oxidative stress develops inside the body, as a result, various transcriptional mediated passages are activated like JNK, p38, IKKβ or NF-κB. IKKβ and also induces NF-κB for activation. JNK, p38 and IKKβ, serine phosphate is further activated for IRS-1 (insulin receptor substrate-1). NF-κB, on the other side, the expression of iNOS that also activates and also catalyzes the S-nitrosylation of IRS-1. Both S-nitrosylation and serine phosphorylation of IRS-1 are phenomena that involve a suppression of way to suppress the phosphorylation of tyrosine of insulin signaling pathways, which ultimately leads to insulin resistance being entered into the liver, adipocytes and skeletal muscles. JNK and IKKB get inspired by oxidative stress and because of metabolism which has a major role in the NF- κB activation and this weaken signaling pathway of insulin which finally leads to the increment of insulin resistance (Figure 5). Glucolipotoxicity and Insulin resistance: Glucotoxicity and lipotoxicity are the most common term refer to glucolipotoxicity. These terms are responsible for the stimulation of pro-inflammatory mediators by activating them and their release which results in an increment of tissue-specific IR and insulin secretion impairment from pancreatic β-cells islets (Figure 6) [99]. Mechanism of Hyperglycemia and Dyslipidemia-Induced Inflammation for the Development of Ir and T2dm (figure 6). The pro-inflammatory mediator's activation gets provoked by Hyperglycemia and Dyslipidemia by the action of various metabolic pathways. These pro-inflammatory mediators induce tissue-specific inflammation once they get released and ultimately result in impairment of insulin secretion from pancreatic islets which results in T2DM [100].
Figure 6. Mechanism of hyperglycemia and dyslipidemia induced inflammation for the development of insulin resistance and T2DM [99].
Role of Mitochondria Mitochondria which arise from the anterior cells of bacteria capable of producing energy in the form of ATP, mitochondria provide a platform for the production of the energy currency of the cell. ATP is essential for many cellular processes. The mitochondria function (and mitochondria dysfunction) plays an important role in metabolic health and cellular destiny. But for the review, we have focused on the role of mitochondria in processes of metabolism including oxidative formation and substrate oxidation (Figure. 7). The rules of the mitochondria function are still complex and it is not fully understood [101].
Figure 7. A diagrammatic representation of the mechanism of mitochondria dysfunction [101].
Mitochondrial dysfunction The alteration in mitochondrial function takes place at the mRNA level (targeted PCR or in more global microarray approach), changes in protein level [99-102] or mitochondrial oxidation [99-106] were made through enzymatic activity and estimation of shape and size of mitochondria (by electron microscope) and substrate oxidation [103, 107]. Accordingly, "some groups indicate the scarcity of mitochondria in terms of mRNA." while others focus on various aspects such as reactive oxygen species (ROS) production. In this review, we mention the word "mitochondria as a reduction in mitochondria the oxidation of substrates including lipids and carbohydrates, resulting in a normal reduction in oxidation phosphorylation. Mechanism of mitochondria dysfunction: In figure (7), Glucose and fatty acid via cell membrane transporters enter the cell. The fatty acid can either be converted into "inert" (TAG) lipid species and "active" (DAG and ceramide) or can be trapped into mitochondria for oxidation in acetyl CoA. as such, glucose metabolized into acetyl-CoA in mitochondria. During the donation of electrons for subsequent ATP repetition in the electron chain superoxide (NADH and FADH2) occurs during electron transport superoxide (02% K) intensifies oxidative stress and the potential induction of NRF2 and the enables activates antioxidant elements to reduce oxidative stress levels [101]. Acetyl-CoA in the citric acid cycle generates NADH and FADH2 which donates electrons for ATP formation in ETS. Superoxide is also formed which leads to development of oxidative stress and NRF2 induction thus activating the antioxidant response to reduced oxidative stress levels [101]. On literary criticism, it can be said that insulin resistance is the product of mixed factors. Insulin resistance increases the demand for a higher beta-cell activity for insulin release, increasing the probability of cell dysfunction and failure. There is a surpassing appeal for beta-cell function in case of insulin resistance that raises a scenario of cell dysfunction and plunge. The streaks of mechanisms result in hyperglycemia or glucotoxicity, lipotoxicity, inflammation and autoimmunity, adipokines, islet amyloid, incretins, and insulin resistance. The exact mechanism of resistance is not clearly defined but it is postulated or different theories that beta-cell exhaustion may lead to hypersecretion of insulin. There is an intensive need for novel approaches to minimize insulin resistance in diabetes. Many therapeutics and controls are available for treatment purposes still a lot of new researches are required for prophylaxis of this disorder. The development of IR by beta-cell dysfunction, oxidative stress, inflammation, physical inactivity, dyslipidemia, JNK and IKKβ, hyperglycemia, genetic factors, and mitochondria dysfunction. Oxidative and metabolic stress can cause oxygenation in various regions. Once insulin resistance has developed, it increases the oxidative stress in the pancreatic beta-cells and peripheral tissues, which reduces insulin secretions and produces insulin sensitivity in the beta cells of the pancreatic islets and peripheral tissues. Much medical and control are available for treatment and still, new researches are needed for prophylaxis of the disease. Besides, we also present a summary of data on various schemes involving insulin resistance.
Conclusion: Diabetes is most frequently known as diabetes mellitus. Diabetes is signalized by immedicable hyperglycemia with most ordinary long-term diabetes associated with damage and dysfunction and failure of different organs, particularly the eyes, blood vessels, hearts, nerve, and kidney. Due to insulin resistance, the increase in the demand for the high beta-cell function to release insulin leads to cell dysfunction and the chances of failure. Factors related to beta-cell dysfunction are hyperglycemia or glucotoxicity, lipotoxicity, inflammation and autoimmunity, adipokines, islet amyloid, incretins, and insulin resistance. There is an intensive need for novel approaches to minimize insulin resistance in diabetes. The mechanism is not understood but it is said that the higher concentration of fatty acids metabolites activates a cascade of serine kinase which intern alters the signaling process. IR is developed by oxidative stress, inflammation, beta-cell dysfunction, physical inactivity, dyslipidemia, JNK and IKKβ, hyperglycemia, genetic factors, and mitochondria dysfunction. Insulin sensitivity starts to fall due to free fatty acids in the case of type 2 diabetes. There is an intensive need for novel approaches to minimize insulin resistance in diabetes. Although prophylaxis of the disease is quite advanced, there is a greater need for research on herbal drugs for diabetes and the use of advanced medical equipment. The main causes of the dysfunction of beta cells are hyperglycemia, lipotoxicity, inflammation and autoimmunity, adipokines, islet amyloids, incretin, and insulin resistance. The mechanism is not understood but it is said that the higher concentration of fatty acids metabolites activates a cascade of serine kinase which intern alters the signaling process. In this article, we briefly mention how the inflammatory mediator, oxidation stresses, the energy mediated molecular and metabolic systems are these subjects. Besides, based on a recent investigation, we have also indicated that the replication of these inflammatory reactions has brought about through a more recent investigation. IR is one of the best ways to prevent pathogen. IR plays an important role in the development of the pathogen and the development of T2DM and the associated complications. Various causes of the development of IR and the finding described here are as follows. The main concern of insulin resistance is that IR is concerned with the activation of many metabolic or intermediate pathways, the provocative and oxidational pro-tension of the intermediaries. Schemes of anti-inflammatory treatment based on the findings described in the above sections are one of the best options to prevent the pathogenesis of IR. However, research carried out to determine the role of anti-inflammatory strategies to prevent IR are still being initiated and needs to be further focused on better and improved diagnostic results in the future. List of Abbreviations: IRS = Insulin receptor substrate PI3 KS = Phosphoinositide 3 kinase P7056 = Ribosomal protein 56 kinase Beta1 mTOR = mammalian target of rapamycin Akt or PKB = Protein kinase B TNF = Tumor necrosis factors α and β JNKS = Jan N – terminal kinase IKK-β = I kappa B kinase β PKC = Protein kinase C ATP = Adenosine triphosphate NF-κB = Nuclear factor kappa-light-chain-enhancer of activated B cells iNOS = inducible nitric oxide synthase ROS = Reactive oxygen species TAG = Triglycerides DAG = Diacylglycerols CoA = Coenzyme NADH = Nicotinamide adenine dinucleotide FADH2 = Flavin adenine dinucleotide NRF2 = Nuclear factor erythroid 2-related factor 2. Conflict of interest: The author expresses no conflict of interest. Acknowledgment: We are genuinely thankful to the department of Pharmacy, Pranveer Singh Institute of Technology, Kanpur, Uttar Pradesh, India, for immensely guiding us and helping while writing this review article. References
|