Pharmacophore an International Research Journal
EPIGENOME ENGINEERING: UNDERSTANDING, MANAGING, AND IMPROVING TECHNICAL ASPECTS
Ahmad Mohammad Khalil1*
|
|
|
ABSTRACT
An epigenomic analysis is big data science, presenting tremendous challenges in its translation into knowledge. To exert precise spatiotemporal control of gene activation and repression, one must have a thorough grasp of the molecular building blocks of epigenetic processes. Only lately has the technology become available to adequately investigate the functional impact of intricate epigenetic pathways. This technology uses a combination of nuclease-null genome-editing (GE) systems and effector domains. Modern epigenome editing (EpGE) modular systems can be adapted to permit precise manipulation of epigenetic marks with no changes in underlying DNA sequence. This review briefly presents the currently achievable epigenetic manipulations along with matching applications in human food and health. The emerging era of Clustered regularly interspaced short palindromic repeats (CRISPR) EpGEs could perhaps be a game-changer for the management of chromatin and epigenetic signature an appealing strategy for therapeutic and breeding purposes. This nascent field is still suffering certain pitfalls.
Keywords: Epigenetics, Epigenome, Genome, Genomics, Gene expression
Introduction
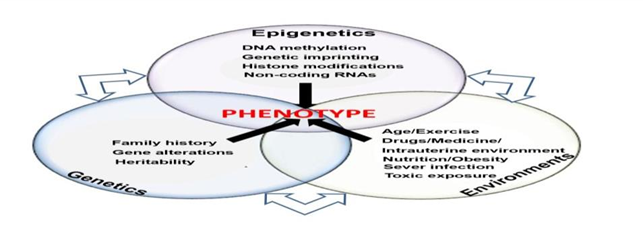
The term “genome” refers to all the genetic material for an organism including humans. On the other hand, the term “epigenetics”, is a literal translation of “on top of the genetics”. Fundamentally, the science of “epigenomics” refers to the record of the modifications in the nuclear architecture, DNA, and histone proteins of an organism. It focuses on examining the impact of chromatin topology, including higher-order chromatin folding and association with the nuclear matrix, DNA wrapping around nucleosomes, covalent modifications of histone tails (acetylation, methylation, phosphorylation, and ubiquitination), and DNA methylation (DM), on a cell's genetic makeup. The concept of epigenetics evolved with time and recent investigations at the molecular level mainly covered non-coding RNAs (ncRNAs) (Figure 1).
|
|
|
Figure 1. Mutual interaction of genetics, epigenetics, and environments to produce a normal or an abnormal phenotype. (Adapted from [1, 2]) |
These modifications occur typically at multiple sites collectively known as histone post-translational modifications. Added to this are the modification of DNA bases in some CpG-rich regions, and the expression of regulatory ncRNA molecules. In recent years, scientists made great steps in clarifying the functional roles of epigenetic processes.
Variations in the structure of chromatin may result from changes to the epigenome, which may then influence how the genome functions [3]. The epigenome controls how genes are expressed, how tissues develop, and how development proceeds. In contrast to the genome, which is kept generally fixed within an individual, the epigenome can be dynamically affected by environmental conditions.
Researchers have identified three different types of epigenetics [4] based on their spatial and temporal properties: a direct form of epigenetics (DE), two forms of indirect epigenetics (within indirect epigenetics (WIE) and across indirect epigenetics (AIE), and a third kind of indirect epigenetics. All of an individual's epigenetic changes that take place throughout the course of their lifetime are categorized as direct epigenetics. The same experience becomes an indirect environmental trigger for the ontogenetic growth of the new person when an epigenetic alteration caused by a direct experience is passed down to the offspring [4]. The whole set of synchronous epigenetic changes that affect the developing person is included in the WIE. Temporarily, it starts when the zygote develops and its surroundings start to change.
According to this fundamental tenet, environmental changes happen simultaneously with the existence of the proto-individual. Indirect epigenetics, which spans from the moment of conception back to the parents' and even grandparents' former lives, is the study of what happens [4]. The epigenomic changes may or may not be heritable. They can be passed down, independently of DNA sequence, from one generation to another through transgenerational stranded epigenetic inheritance. This mode of inheritance refers to the transmission of epigenetic tags, through generations, that influence the traits of offspring without changing the primary DNA structure [5]. Due to the action of the maintenance DNA methyltransferases (DNMTs), a type of post-replication alteration called DNA methylation (DM) can be passed down via cell division [6]. These enzymes create 5 mC by transferring a methyl group from S-adenyl methionine to a cytosine ring’s fifth carbon. Demethylation, the opposite process, can be passive or active and is more difficult. TET enzymes oxidize 5 mCs and aid in the elimination of DM from a specific locus [5, 7]. The methylation of DNA and histones induces down-regulation of gene expression, while histone acetylation and phosphorylation enhance gene expression [8]. According to yeast the memory of DM may last for millions of years [5], indicating that daughter cells may get a gene expression profile from parental cells (a process known as "cellular memory") or at the generational level when a parent passes on a trait to its children.
Biotechnologies that attempt to modify endogenous, site-specific epigenetic targets are responsible for this advancement. Despite recent progress, significant work has to be done to fully understand how epigenetic pathways affect cellular programming, illness, and tailored therapy.
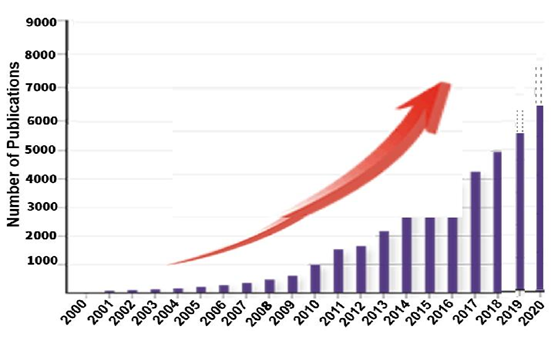
The speed of epigenetic research picked up drastically from the early 2000s. Figure 2 demonstrates that between 2000 and 2010 the number of published articles found in the database that had the word ‘epigenetics’ in their title grew up to 1000. The public prominence of epigenetics increased with the launching of the Human Epigenome Project. By 2015, the main information about functional basics that control gene expression in about 127 human tissues and cell types was disclosed. By that time the figure of available research papers jumped to about 3000. Following this, the number of published epigenetics papers mushroomed, so it is hard to keep up. Numerous diagnostic and pharmaceutical corporations were also developing tests and candidate epi-drugs [7]. Just how far the area progressed can be realized from the statistics that in 2016 experts from Grand View Research forecasted that the global business market for epigenetic products would reach USD 16.31 billion by 2022.
|
|
|
Figure 2. Search results of epigenetic publications per year by each keyword in PubMed and the database of ISI Web of Knowledge. |
Materials and Methods
This survey was based on screening the recent literature up to July 2023 using PubMed and the database of ISI Web of Knowledge. The keywords used for the search were (“Epigenetics”), (“Epigenome editing”), (“Genome editing”), (“Functional genomics”), (“Gene expression”), and (“CRISPR/Cas9”). After the exclusion of duplications and non-English publications, a total of 80 articles were selected. These papers were analyzed and reviewed to (1) list and define the EpGE architecture and approaches, (2) assess the applications of EpGE, and (3) present the challenges of EpGE and suggest alternate solutions. Only a single 2007- paper and another in 2010 were used to lay down the EpGE approaches, and the mechanisms regarding gene expression and regulation. There were 21 published between 2011 and 2018. Fifty-seven of the examined research works appeared during the years: 2019, 2020, 2021, 2022, and 2023.
Results and Discussion
The fundamentals of DNA-binding are the same in both GE and GE processes. However, EpGE techniques lack the endogenous DNA repair mechanisms that GE procedures provide. In contrast to GE, which modifies the actual DNA sequence itself, EpGE modifies and displays DNA sequences for proteins and other DNA-binding substances that influence DNA function.
Conceptionally, EpGE utilizes the two main features of epigenetic marks: heritability and reversibility [9]. The first means that the epigenetic mark of DM and certain histone modifications are passed over generations. The second means that the cell can respond to external stimuli by adjusting these marks. The Epigenetic enzymes are considered the effectors of both maintaining and modifying epigenetic signatures. The proper epigenetic mark(s) assigned to effectively modify gene expression should be confirmed for any given chromatin context and must be (mitotically) stable. Rewriting an epigenetic mark can be done by epigenome editing to change how an organism's endogenous genes are expressed. A catalytic domain (CD), also known as an effector domain (ED), writer, or eraser, is directed to a particular locus with the goal of rewriting a gene's epigenetic imprint. The ED is fused to a programmable gene-specific DNA-binding domain (DBD) in order to do this; a linker peptide is employed to join a DBD and an ED. Writers are the enzymes in epigenetics that add the moiety to histone tails. Erasers delete epigenetic changes on a specific basis. In order to guarantee the existence of the proper histone modification to manage the required transcriptional program, writers and erasers coexist in harmony [10]. Dysregulation of one or both causes aberrant histone modifications, which alter transcription. A CD that allows the enzymatic reaction of moiety transfer to occur is required for all epigenetic writers, as is another domain that allows the identification of chromatin. For instance, HMT (the writer) transfers a methyl group onto definite lysine residues on histones. On the other hand, the Jumonji domain-containing histone demethylase (eraser) removes histone methylation by oxidation reactions.
Updated History of Epigenome Editing
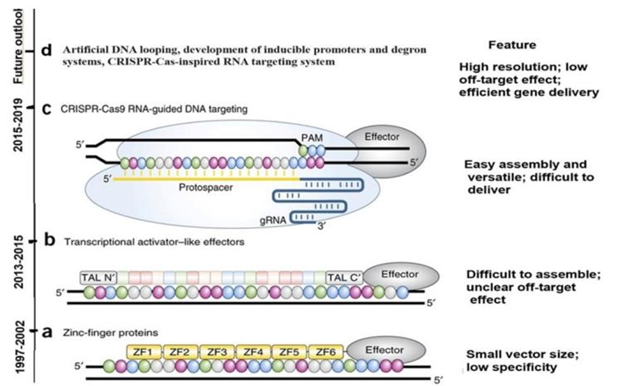
Over the years, various EpGE tools were utilized to change DM at predetermined sites (Figure 3). With the help of the catalytically dead (CRISPR) associated Cas9, also known as dCas9, several research teams [11-13] have advanced technologies for targeted gene expression. These technologies include the zinc finger (ZF) and transcription-activator-like effector (TALEs) fusions. Since the CRISPR-dCas9 system is an RNA-guided DNA-targeting system, it has several advantages over ZFs and TALEs in terms of targeted gene activation.
This eliminates the requirement for intricate protein engineering and may create new opportunities for spatiotemporal control of gene expression. Researchers demonstrated the efficacy of using epigenetic effectors connected to DNA-targeting proteins to modulate epigenetic landscapes in addition to transcriptional activators for directed gene expression.
However, compared to TALE-based activators, several results revealed that dCas9-based activation tools cause lower amounts of transcription [12].
|
|
|
Figure 3. The evolution and application of epigenome editing tools. (a) The first site-specific DNA methylation tool was introduced in 1997. In this in vitro procedure, a bacterial methyl transferase (ED) was fused with ZF proteins (ZFPs) to identify a nine-nucleotide DNA sequence. In 2002, a ZEP was also attached to CDs of HMT to edit histone 3 methylation of Lysine 9 (H3K9) methylation in vivo. (b) Then the CDs of DMT3a and Ten-eleven translocation hydroxylase or Lysine-specific demthylase 1 were fused TALE. (c) The dCRISPR-associated (Csas9), or dCas9, enormously extended the EpGE toolbox during the period 2015 to 2019. (d) Prospective expectation. (Adpted from 15, 16) |
It was reported in 2015 that the p300 HAT CD has been modified using CRISPR-dCas9, TALE, and ZF fusions [13]. In order to cause the transactivation of genes, this places histone 3 lysine 27 (H3K27) and additional acetylation sites on histone tails (Table 1). In order to activate gene expression, other epigenetic effectors that catalyze covalent modifications in DNA have also been linked to DNA-binding proteins. As an illustration, TET demethylase enzymes were used with ZFs [14], TALEs [11], and CRISPR-dCas9 [15] to direct promoter-specific DNA demethylation and, consequently, upregulate gene expression. These enzymes catalyze the sequential oxidation of 5-methyl deoxycytosine (m5dC) to form 5- hydroxymethylcytosine, 5-formyl cytosine, and 5-carboxylcytocine.
Programmable Epigenome Editing Systems
Currently, three major sets of DNA-targeting proteins are largely employed in the EpGE system (Table 1) ZFPs), TALEs [16], and CRISPR [3, 17]. Epi-effectors consist of intended DNA recognition domains (ZF, TAL effector, or adapted CRISPR/Cas9 complex) and CDs from a chromatin-adjusting enzyme [18]. The CRISPR/dCas9 system is now the most extensively used platform because it takes less time and money to modify and implement the technology and because it is very effective and adaptable [19, 20].
Table 1. Summary of effector protein domains selected for epigenome editing systems and their effects on gene expression.
|
Chromatin/DNA Targeting Module |
Effector Protein Domains* |
Effects/Applications |
|
ZFP |
M. SssI (bacterial DNA methyltransferase) |
DNA methylation in vitro |
|
SUV39H1 and G9A catalytic domains (histone methyltransferases) |
Histone H3K9 methylation at the VEGF-A locus in cells |
|
|
G9A catalytic domain (histone methyltransferase) and p65 of NF-kB (to induce histone acetylation) |
Histone modifications at FosB locus in nucleus accumbens of mouse brain |
|
|
Full length or the self-association domain of Ldb1 (transcription cofactor) |
Induce chromatin looping at β-globin locus or fetal γ-globin locus |
|
|
TALE |
TET1 catalytic domain (Ten-eleven translocation methylcytosine dioxygenase) |
DNA methylation in vivo |
|
DNMT3a catalytic domain and DNMT3l (mammalian DNA methyltransferase complex) |
DNA methylation of CDKN2A gene in vivo |
|
|
LSD1 (histone demethylase) |
histone H3K4 demethylation at multiple enhancers in vivo |
|
|
SpdCas9 dCpf1 SpdCas9 SpdCas9-SunTag |
VPR VPR or p65 VP64 (VP16 tetramer) VP64 (VP16 tetramer)
|
Transcriptional activation Transcriptional activation Transcriptional activation Transcriptional activation and cellular reprograming Can recruit p300 to deposit H3K27ac |
|
SpdCas9 |
KRAB Mxi1 SID |
Repression of transcription by blocking transcription complex formation. May also recruit other repressive modifications |
|
SpdCas9 SpdCas9 SpdCas9-SunTag |
DNMT3A full-length DNMT3A catalytic domain DNMT3A full-length or catalytic domain |
Repression of transcription by DNA methylation in vivo |
|
SpdCas9 SpdCas9-SunTag SpdCas9 |
TET1 catalytic domain TET1 catalytic domain TET1 catalytic domain |
Activation of transcription by DNA demethylation in vivo |
|
SpdCas9 SpdCas9 |
G9a (EHMT) SUV39H1 catalytic domain |
Repression of transcription by targeting H3K9me |
|
SpdCas9 |
LSD1 |
H3K4me2 demethylation Causes H3K27ac removal Both repress transcription. Ideal for enhancers |
|
SpdCas9 SpdCas9-SunTag |
p300 catalytic domain p300 catalytic domain |
Targeted H3K27ac Ideal for enhancers |
|
dCas13 dCas13 |
ADAR2 Modified ADAR2 |
RNA editing Conversion of adenosine to inosine Conversion of cytosine to uracil |
*Effector domains that interact with dCas9 can be divided into two categories: (i) transcriptional regulators that sequentially Use other proteins, such as transcriptional cofactors, chromatin modifying enzymes, and chromatin remodeling complexes; and (ii) chromatin modifying enzymes as such that catalyze the removal or addition of epigenetic marks. For instance, two effective transcriptional regulators, the Krueppel-associated box (KRAB) domain and the herpes simplex VP16, or the substitutes, have been employed successfully to activate and inactivate gene expression, respectively. To add or delete chromatin marks from histones or DNA, a variety of chromatin modifiers were used. The development of the dCas9-SunTag system made it possible to simultaneously enroll several copies of the same effector or other transcriptional regulators at a target location. The creation of dCas13 enabled the control of RNA in addition to DNA and chromatin editing techniques. Histone acetyltransferase, P300 ADAR: RNA-specific adenosine deaminases; catalytic domain (CD); DNA methylation is DM. DNA methyltransferase (DNMT); HMT: histone methyltransferase; H3K4: lysine 4 methylation on histone 3; H3K27: lysine 27 methylation on histone 3; TA stands for transcriptional activation, while SpdCas9 stands for Streptococcus pyogenes dead Cas9; TET1 stands for ten-eleven translocation methylcytosine dioxygenase 1, VP16 stands for herpes simplex virus protein 16, and VP64 stands for four tandem copies of VP16. (Adapted from 15, 18])
CRISPR-Cas systems are DNA-encoded and RNA-guided, in contrast to ZF- and TALE-based systems, which rely on protein-DNA interactions to target precise genetic loci [21, 22]. By attaching epigenetic effectors to the dCas9 protein and focusing on regulating regions like promoters and enhancers, epigenome editing has proven to be more precise and effective [23]. Because they can avoid the need for intricate protein engineering to facilitate DNA detection, CRISPR-Cas systems are given significant advantages by this property [24]. Gene-targeted epigenome reprogramming has received a lot of attention since the invention of CRISPR/Cas [25]. The trans-activating CRISPR RNA (tracrRNA) and the CRISPR RNA (crRNA) form a chimera with the Cas9 nuclease in the well-studied type II CRISPR system. Single-guide RNA (sgRNA) is a common name for this hybrid [26]. The NGG-trinucleotide repeats, which are located upstream of a protospacer adjacent motif (PAM), are complementary target DNA sequences that the sgRNA uses to activate dCas9. The CRISPR/dCas9 system may be used to target practically any genomic location of interest by changing the sequence of the gRNA [27]. It's interesting to note that numerous loci can be addressed equally, allowing for efficient multiplexing. CRISPR-Cas9- based EpGE uses an engineered SpCas9 variant, which is purposefully stripped of its catalytic activity into a catalytically “dead” or deactivated Cas9 (dCas9). The dCas9 system was used to target epigenetic reprogramming to initiate site-specific DM. By merging the DNMT3a CD with the dCas9 protein, dCas9-DNMT3a can achieve targeted DM of a targeted zone as dictated by the present sgRNA [25, 28]. Likewise, dCas9 was combined with the catalytic core of the human acetyltransferase p300 (Table 1). The dCas9-p300 complex effectively catalyzes targeted acetylation of lysine 27 in histone H3 (H3K27) [29].
While a dCas9 fusion to a repression domain demonstrated the CRISPR-dCas9 system's capacity to use a heterochromatin-forming complex to mute gene expression, binding of dCas9 to an epigenetic effector that acetylates histone tail residues leads to transcriptional activation. This system exerts a positive or negative control on the expression of developmentally relevant genes, which can govern specific differentiation decisions and direct the differentiation status of the cell down desired pathways. It is possible to utilize CRISPR in conjunction with a dCas9 fusion protein to reversibly modify DNA and heritably suppress the expression of "most genes" [30, 31]. Generating versions that can target multiple effector domain copies to a single site and targeting epigenetic alterations that, in certain situations, may be transferred to the next generation without the targeting assembly are recent breakthroughs in evolving CRISPR-dCas, systems [22, 31]. Combining effector domains and targeting strategies to produce synergies that improve the system's performance. A method established to manipulate the differentiation state of human pluripotent stem cells (hPSC) [21] is another aspect of this. A CRISPR EGE variation known as FIRE-Cas9 enables undoing the alteration if something goes wrong.
Similar to how genes are activated, repressor domains, or epigenetic effectors, are combined with DNA-binding proteins to silence endogenous gene expression at specific loci. The KRAB repressor domain, which is highly conserved, is the one that is most commonly used to silence genes (Table 1). Through interactions between the KRAB-associated protein 1 corepressor and other factors that catalyze histone methylation and deacetylation, complexes that result in the development of heterochromatin is recruited, which facilitates KRAB repression. Strong inactivation from the promoter and from the proximal as well as the distal regulatory elements has been demonstrated by the attachment of KRAB to ZFPs [3], TALEs [3], and CRISPR-dCas9 [32].
Researchers have discovered that additional repressor domains, such as the major interaction domain (SID) [3] and Mxi1 [9], can effectively silence genes in addition to the KRAB repressor. When dCas9-KRAB and dCas9-Mxi1 were examined, it was shown that the Mxi1 fusion may cause roughly three times as much repression [9]. According to different research [3], TALE-SID repressors may suppress transcription 26% more than their TALE-KRAB counterparts. Additionally, four SID (SID4X) domains concatenated together and bound to DNA-binding proteins can provide greater repression than a single SID domain fusion [3]. This is comparable to the connecting of ZFPs and their VP64 tetrameric form to VP16 units, which are involved in the enrolment of chromatin-modifying factors that improve chromatin accessibility [3].
Epigenetic effectors may accurately catalyze particular alterations in histones and DNA that result in targeted epigenetic repression, in contrast to transcriptional repressors, which adequately attract heterochromatin-forming complexes to desired loci. At target promoter and enhancer zones, histone demethylases [3, 32], HMT [32], as well as DNMTs [15] with ZF, TALE, and CRISPR-dCas9 proteins showed substantial suppression efficacy rates.
RNA alterations have recently emerged as key posttranscriptional regulators of gene expression strategies [33]. Understanding the functional significance of RNA changes in regulating coding and non-coding RNA processing and function, which in turn systematically shapes certain gene expression patterns, is making significant strides. They have an impact on several biological functions, and many of these alterations must be precisely deposited in order for growth to proceed normally. The modulation of circadian rhythms, spermatogenesis, embryogenesis, DNA damage, stress response, pluripotency, and cell reprogramming are only a few of the physiological processes that m6A deposition in RNA is essential for [34]. The N6-methyladenosine (m6A) regulators are also tightly connected to either oncogenic or tumor-suppressiveactions [35].
Applications
EpGE, utilizing an inducible mechanism, offers a wide range of possible applications in addition to being a formidable tool for addressing fundamental epigenetic concerns in basic research. It aims to hasten the development of solutions and applications for everyday problems, from breeding to medicinal ones [21, 35, 36]. In recent years, bioengineers have developed effective metabolic pathways for ethanol generation in algae or maize, leading to the development of sustainable and affordable sources of renewable energy. They succeeded in improving crops and farm animals, as well as in developing novel treatment approaches for inherited and acquired disorders-associated epi-mutations. In the next sections, two main proofs of concept studies demonstrating the application of EpGE are discussed.
Plant Epigenome Editing
Because the yield and quality improvement of vegetables and crops are attracting attention as they are a source of food security for humanity, research in the field of plant EpGE is appealing and accelerating rapidly. Currently, this technology is opening the door to developing horticulture. It is valuable to explore epimutations in a range of crop species while understanding the basic features of epigenetic regulation of agronomically key traits such as yield, quality, and increase resistance to drought, disease, and to other stress factors [16, 22, 36-40]. Additionally, epi mutagenesis and focused control of transcriptional regulation are employed to increase our understanding of the relationships between critical proteins, alter plant developmental phenotypes, and advance our understanding of how plants accumulate, retain, and utilize DM [22]. These proteins have an impact on several elements of RNA metabolism and other biological processes in plants, such as seed formation, growth of the leaves and roots, floral transitions, and fruit ripening (Figure 4). This involves an apparatus containing “writers” (methyltransferase complex components), separated by “erasers” (demethylases), and documented by “readers” (Proteins that connect with m6A). The readers are corporate with m6A-adjusted RNA and control RNA splicing, RNA stability, and 3' untranslated regions (3′UTR) handling [41].
|
|
|
Figure 4. Control of plant's growth and development by post- transcriptional changes using m6A, the most prevalent internal modification present in the mRNAs of all higher eukaryotes. Post-transcriptional alterations are controlled by diverse writer, eraser, and reader proteins that affect different plant developmental processes such as seed development, leaf, and root growth and floral transitions, and fruit ripening [42, 43]. |
The ZF-based EpGE applied to A. thaliana and O. sativa, as well as the TALE and dCas9-based tools in A. thaliana and O. sativa, respectively, are among the tools employed in plants [41]. Furthermore, a great number of EpGE approaches based on small RNAs are hired for regulating gene expression in A. thaliana Nicotiana benthamiana O. sativa, Solanum tuberosum and Z. mays [22, 39, 44]. For example, reports on the use of the RNA-directed DM (RdDM) containing transgenically generated exogenous small interfering RNAs (siRNAs) in A. thaliana the RNAi-based tools in O. sativa [28, 41]. Moreover, tissue culture-based EpGE DNA and/or histone modifications were accomplished in Caribbean agave angustifolia), Henequen (A. fourcroydes) A. thaliana, common tobacco (N. tabacum) O. sativa Pinus radiata Z. mays [44].
In addition to utilizing activator domains to directly activate transcription, another option is to use domains that can add active or remove restrictive epigenetic markers, indirectly activating gene expression. For instance, the p300 domain from humans have been used to target transcriptional activity in plants through H3K27 acetylation. Another method is to employ the CD of the plant-specific Arabidopsis HAT of the cAMP-responsive element-binding protein (CBP) Family 1 (HAC1). Although HAC1 can increase the transcription of targeted genes, using VP64 results in a higher amount of activation, at least for the gene p300 [45]. A highly efficient tactic is the combination of many distinct activator domains to cooperatively trigger gene expression [45, 46]. The stimulation of gene expression was boosted by VP128 to VP128 to TAL activator domains being targeted directly. Additionally, the combination of VP64 with P65 and Rta (Table 1), two additional effectors domains that had previously been demonstrated to function in animals (VPR), was similarly capable of significantly increasing the amount of gene expression in plants compared to VP64 alone [45, 46].
For targeted gene repression in plants, the most frequently used EDs are the Arabidopsis ethylene response factor- associated amphiphilic repression domains, like those found in SUPERMAN or BODENLOS. These domains are used widely as a means to study highly redundant genes, since these repressor domains override activator domains, including VP16, and can be employed to turn a constitutive activator into a constitutive repressor [47]. Targeted addition of DM to promoters utilizing DNMTs is used fruitfully to repress transcription. For instance, the CD of the RdDM-based DNMTs from tobacco (N. tabacum), Domains Rearranged Methyl Transferase (DRM) can add DNA methylation to a target promoter, leading to transcriptional repression [48].
In addition to altering the epigenome for transcriptional repression, CRISPR interference (CRISPRi) is a direct method of suppressing gene expression by impeding RNA polymerase II activity. The use of CRISPRi has only been reported in plants, and only one instance of maize (Z. mays) has shown that CRISPRi can partially suppress a gene [43]. There are no studies on co-targeting several repressor domains to suppress transcription in plants [22].
Epigenome Editing in Medicine
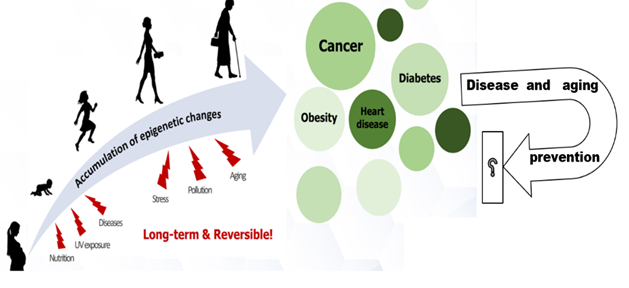
Accumulating evidence proved that, relative to normal tissues, changes in the genome (genetic mutations), changes in the epigenome (epi-mutations), or both are intimately associated with the development and progression of human diseases, including cancers, type II diabetes mellitus, brain disorders, cardiovascular dysfunction, and even psychological disorders, to mention a few [8, 9, 14, 49, 50]. The epigenome, which serves as a link between the environment and the genome, is dynamically altered throughout the onset and development of many different diseases (Figure 5). By epigenomic sketching before the onset of diseases, it may become possible to tailor interferences to prevent chronic disorders or diseases in future life. Through the abundance of emerging signals signifying the involvement of epigenetic processes in the usual diseases, researchers tried to trace the epigenomic changes that can switch on or off genes implicated in cell growth or the immune response. Also, they attempted to use epigenetic biomarkers to detect new risk factors that might be linked to disease status.
|
|
|
Figure 5. Environmental factors, acting at various moments throughout the life cycle, can result in epigenetically-mediated alterations in gene expression and consequently in phenotype. Potential public health intervention may become reality. (The idea inspired from: [51]). |
When adequate gene therapies have not yet been developed or are unsuitable, targeted regulation of disease-associated genes may allow for innovative treatments for a variety of disorders [18]. Although population-level and transgenerational consequences are not well understood, they may play a significant role in applied operational genomics and customized medicine [17]. Numerous epigenetic biomarkers and medicines have recently entered clinical trials for a variety of solid malignancies and have recently been licensed by the Food and Drug Administration (FDA) for the treatment of myelodysplastic syndromes and leukemias [35, 49]. It appears that various transcriptional activators produce gene expression at various intensities [3]. For targeted transcriptional activation, Farnesyl pyrophosphate synthase (FPPS) has also been linked to transcriptional activator domains, such as the p65 component of the nuclear factor kappa B complex (Table 1). A well-known therapeutic target, the enzyme FPPS, is located in the mevalonate pathway. The subject of research is a recently discovered druggable pocket adjacent to the enzyme's active site [52]. Pharmacological modulation of this compartment is seen as promising, although its potential biological role is unknown at this time.
By concentrating on the maspin promoter region, researchers were able to revive a dormant mammary serine protease inhibitor (maspin) tumor suppressor gene [53]. In aggressive epithelial cancers, this gene is frequently epigenetically silenced. The ZFPs EpGE still has a bright future in treating many neurodegenerative illnesses [54]. A target gene's expression was reported to be boosted by five times or more when TALE activators were directed to the promoter region of the endogenous human vascular endothelial growth factor-A gene, which is up-regulated in many cancers and contributes to tumor angiogenesis [11]. Similar results showed that TALE fusions with the transcriptional activator VP64 boosted the endogenous levels of gene expression of a few human pluripotency factors by a ratio of two to five (Table 1). Combinations of TALE-VP64 fusions that target the promoter sites of genes associated with inflammatory, immunomodulatory, and cancer-related pathways established the presence of synergistic activation action at target areas, providing opportunities to build adjustable transcriptional networks. [11].
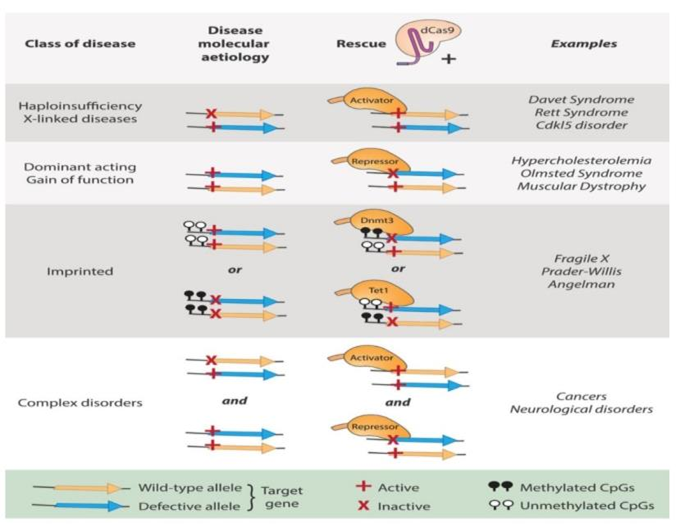
Figure 6 lists some diseases that would make good candidates for the development of innovative epigenetic therapies. Neurological illnesses may benefit from epigenetic editing. One of them is the fragile X syndrome, the most common kind of intellectual impairment in men, in which aberrant DNA methylation causes the FMR1 gene in neurons to be epigenetically silenced. Induced PSC-derived neurons once again expressed FMR1 after a precise demethylation of the FMR1 promoter using the dCas9-TET1 system flipped its heterochromatic status [55]. Another instance of a similar approach being successfully used to treat imprinting diseases, Prader-Willi (PWS) and Angelman (AS) syndromes, which are brought on by an abnormal DM fashion, was described [19].
|
|
|
Figure 6. Prospective therapeutic applications of EpGE. Different categories of disease could benefit from the development of discrete CRISPR/Cas9-driven EPGE strategies. Examples of how mutant or wild-type alleles could be maneuvered in specific disease contexts are depicted here. The molecular etiology underlying each disease class, as well as the dCas9 -based rescue strategy, are present [19]. |
Targeting (i) cytokines in the inflammatory pathway involved in treating degenerative disc disease [56] or (ii) cutaneous pain receptors [57] can be used to treat a variety of inflammatory and pain-related disorders. Surprisingly, because Olmsted syndrome and other autosomal dominant diseases are connected to the skin, it makes the skin an accessible and appealing organ for CRISPR-mediated gene silencing.
It was reported on the molecular basis for m6A deposition in RNA, variations in this deposition, biological function, and implications in a number of disorders, including cancer. It was also discussed how the presence of m6A, m5C, and pseudouridine (Ψ) in coding and non-coding RNAs plays a physiological and pathological function [32]. Changes in tRNA modifications, such as m5C or 5-methoxycarbonyl methyl uridine (mcm5U), have also been linked to cancer and are indicative of altered protein translation [58].
EpGE, like RNA editing, excludes genetic modifications and the hazards connected with them [22]. Using CRISPR- dCas9to suppress the expression of the gene SCN9A, which in human codes for a sodium ion channel, is an illustration of a potential functional use of EpGE.This approach revealed therapeutic possibilities in three mouse models of chronic pain [59]. Since the link between epigenetic alterations, their effects on gene expression, and the knowledge that these epigenetic changes are reversible have been established, new therapeutic avenues for the treatment of many diseases, including diabetes, cancer, and neurodegenerative illnesses, have become available [24].
Recent research [60] expressed its effectiveness in decreasing the levels of tau protein, a group of six protein isoforms involved in maintaining the stability of microtubules in the axons of the central nervous system [61]. These proteins control a protein involved in Huntington's disease, targeting an inherited form of obesity, and Dravet syndrome. It is understood that cancer is correlated with epigenetic changes, and hence CRISPR/Cas system displays capacity for cancer therapy using EpGE [8, 24, 54, 62]. By simultaneously turning on tumor suppressor genes and suppressing the production of oncogenes, this objective can be accomplished [63]. In this regard, simultaneous activation and repression of several genes in the same cell were achieved in vitro by combining dCas9 with chemically and photochemically inducible effector proteins [59] and by creating platform gRNA molecules that may use transcriptional regulators [59].
Limitations
Up until recently, comparative EpGE methods have shown a strong correlation between epigenomic traits and genome regulation. They do not, however, differentiate between the amount to which epigenetic circumstances actually cause gene activity states as opposed to merely correlating with them. These methods not only point out the drawbacks and technological restrictions but also imply that epigenetic markers do not have clear-cut purposes. Sequence specificity is decisively vital in EpGE therefore it must be wisely confirmed. EpGE faces the challenge of standardization for every new product, including finding an appropriate transformation vector and cloning the editing constructs with suitable genes and promoters. A significant limitation of the potential for EpGE as a biomedical plan in humans is the delivery of the system into the target cells. Several vehicles for the delivery of the large dCas9 fusion proteins to different types of target cells have been acknowledged [20, 26, 56, 64-69]. However, obstacles still prevent the widespread use of these tools for in vivo investigations. For example, delivery of the large dCas9 fusion proteins to different types of target cells and tissues is a hurdle to extensive implementation of these tools for in vivo studies [20, 26, 56, 64, 65].
Furthermore, it unclear if the TALE combination will have an impact on the epigenome modifier's catalytic activity. In effector proteins that require multiple subunits and multiplexes, this may be very important. The ligands and substrates at the target site maybe blocked by the proteins utilized in EpGE [44]. If they are directed to the same sequence, transcription factors themselves may compete with the TALE protein [51].
The vulnerability of the dCas9based EpGE systems to off-target effects, which can come from a variety of sources [70], is another issue. For instance, using a suboptimized sgRNA and a dCas9 enzyme domain fusion to initiate EpGE. The chromatin alterations might also be reversed by DNA repair mechanisms, preventing the desired modifications from developing. In plants, strategies related to the reduction of off-target mutations and the decrease in generation time also are important. Elimination of the editing reagents in plants may take manifold generations and therefore the developments in temporary expression are desirable. The accurate mapping of m6A changes at the level of cells may be aided by a variety of radical sequencing methods, including MeRIP-seq, immunoprecipitation, and miCLIP (m6A individual-nucleotide-resolution cross-linking) [49].
Conclusion
The ease with which GE and EpGE may be carried out has been significantly enhanced with the discovery, description, and adaptation of the CRISPR-Cas9 system. EpGE, facilitated by CRISPR/dCas9, develops a very effective targeted method for potential therapeutic applications in precision medicine. The CRISPR -Cas9 technology may be used in new ways to enable EpGE applications. Targeted gene activation or repression was made possible by the fusing of chromatin-modifying domains of dCas9 in both cultivated cells and in vivo animal models. Nevertheless, despite the substantial progress made in recent years to elucidate the molecular basis of epigenetic processes, EpGE is still a developing discipline that faces several problems and unanswered concerns. These limitations are related to off-target effects, editing efficiency, delivery, cytotoxicity, specificity, and stability of epigenetic states. It is anticipated that a conformationally activated CRISPR/Epi--editor, where the epigenetic enzymatic domains are functionally connected into one of the nuclease’s domains of Cas9, will have more resolution and lower the off-target impact to potentially allow therapeutic uses in humans. Such off-target effects might be reduced by an inducible promoter that controls the interval and level of dCas9 fusion protein production.
In the clinical arena, where epigenomic medicine may have an impact, continuing, pre-clinical, and clinical trials are evaluating epidrugs individually or in combination with other pharmaceuticals. Overcoming the rising resistance and broadening the therapeutic profile outside hematological malignancies are the chief challenges faced by conventional epigenetic drugs. Thus, innovative approaches in the field of epigenetic treatment are urgently needed. Prior administration of an epi-drug, which can increase chromatin openness to chemotherapeutic drugs through chromatin decompaction, was suggested to make cells more sensitive to chemotherapy. The joint use of patient-derived iPSC and EpGE allows accurate disease modeling and investigations of the interconnection that epigenetic marks play in disease progression. Because of the intricacy of interactions, "programable" EpGE technologies are being created that may target or alter the epigenetic state at certain genomic loci, allowing for the analysis of those loci's context-dependent function. Combinatory gene targeting strategies in cancer therapy will take a lot of hard work to develop effective delivery methods that can target the whole population of disease-associated cells. Considering the study that will be conducted in the upcoming years that focuses on scientific advancements in these sectors.
Finally, the discussion would be incomplete without mentioning some moral and ethical concerns that may result from the applications of EpGE. EpGE does not alter the genome directly, although it is currently believed to have less of an effect on germ cells than GE [71-74]. However, because of the probabilistic transgenerational epigenetic inheritance in methylation edited mammals [75-77], several questions and uncertainties about the health, safety, or other impacts of epi–engineered crops and foods have been raised [78].
Will scientists be able to spot epigenetic alterations brought on by possibly dangerous exposures in populations? Would these modifications act as a biomarker or signal of a higher chance of developing an illness in later life? What is the possibility of discrimination against people using social epigenetic data? Worldwide, there are calls for the adoption of policies that comprehensively regulate the use of private epi/genetic information for non-medical reasons by third parties.
Acknowledgments: None
Conflict of interest: None
Financial support: None
Ethics statement: None