Pharmacophore an International Research Journal
MOLECULAR CHARACTERIZATION OF HEMOPHILIA A PATIENTS IN DUHOK, IRAQ
Najeeb Saeed Rasheed1, Adil Abozaid Eissa2*
|
|
|
ABSTRACT
The gene for coagulation factor VIII in hemophilia has more than 250 distinct mutations, and documentation of these mutations can predict the clinical phenotype, the likelihood of developing an FVIII inhibitor, and how an individual will react to the induction of immune tolerance. The current study set out to identify common mutations underlying hemophilia in a single center from Duhok/Iraq. All enrolled patients had molecular studies using inverse shifting PCR (IS-PCR) and limited sequencing to identify hemophilia-associated mutations. The current study included 80 hemophiliac patients with a median age of 15. 77.5% of patients (62) have a family history; the initial clinical features at diagnosis included skin and mucous membrane bleeding. No inhibitor was found among the enrolled patients, and the most common chronic complication included targeted joints in 56 individuals (70%) and predominantly involved the knee, elbow, and ankle joints. Other complications include viral infection, particularly HBV and HCV (8/80, 10%). The most common genetic mutations were Inversion 1, followed by Inversion 22 and point mutations in 35 (43.75%), 32 (40%), and 10 (12.5%) patients, respectively. One novel point mutation in exon 14 and only 3 (3.75%) cases remained uncharacterized. Inv. 1 and Inv.22 were associated significantly with the severity of hemophilia, target joint formation, more bleeding episodes, chronic pain, and viral hepatitis. Genetic heterogeneity of Hemophilia A in the region with predominant Inv. 1 is mainly related to ethnic variations.
Keywords: Hemophilia, IS-PCR, Inv.1, Inv.22, Factor VIII inhibitor
Introduction
Hemophilia A is a genetic bleeding disorder caused by a clotting factor VIII (FVIII) deficiency necessary for normal blood clotting [1]. According to the remaining FVIII activity in plasma, hemophilia A is classified into mild, moderate, and severe disease types, with 5-40%, 1-5%, and <1%, respectively. This classification provides information on different types of bleeding and the incidence of hemorrhagic episodes [2].
The gene of factor VIII in humans is located on the X chromosome and contains 26 exons that span approximately 186 kb of genomic DNA. Mutations in the FVIII gene have been documented to induce the clinical phenotype [3]. In addition to frequent inversions of intron 1 and intron 22 in the FVIII gene, these mutations include numerous deletions, insertions, missenses, nonsense, and splice site changes. [4]. Examining this disorder still includes a crucial and essential role in molecular genetic diagnosis. This information is useful for correlating genotype and phenotype, understanding the basis of inhibitor development, new approaches to treating hemophilia such as developing new clotting factor concentrates and gene therapy, carrier discovery, and prenatal diagnosis while facilitating our understanding of the functional biology of this gene [5].
Direct genetic analysis of hemophilia A disease requires several technical approaches to cover the entire field of hemophilia A defects, thus necessitating numerous expensive and time-consuming procedures [4]. As a brief overview, 25-50% of all hemophilia A cases are caused by an inversion resulting from homologous recombination involving intron 1 or 22 and related sequences outside the FVIII gene [6]. Several methods now frequently used to detect these inversions have been developed, including targeted PCR, Southern blot, Long Range (LR-PCR), and Inverse Shifting (IS-PCR). In order to detect approximately 50-75% of the other mutations affecting the FVIII gene, all 26 exon regions need to be analyzed, including the flanking splice sites and promoter regions, utilizing Sanger DNA sequencing [7].
In recent years, gene dosage analysis using multiplex ligation-dependent probe amplification (MLPA) has emerged as a method of detecting large deletions and duplications; it allows for the simultaneous detection of multiple mutations or deletions in the factor VIII gene. It involves hybridizing a set of specific probes to the target DNA and amplifying the hybridized probes [4]. Next-generation sequencing (NGS) has become a routine diagnostic procedure in several genetic molecular laboratories specializing in hemophilia or other coagulopathies [8]. Also, array-comparative genomic hybridization (a-CGH) enables the detection of gains and losses within genomic regions [9].
As the community in Duhok province, to some extent, is closed, the current study has been initiated to identify mutations commonly encountered among hemophilic patients in the region and to compare their prevalence with other populations from the country and nearby regions.
Materials and Methods
The current study was done to document the molecular basis of all hemophilia A patients registered at the Jin Center for bleeding disorders in Duhok province over 1 year (1st of November 2021 to 30th November 2022). All male hemophilia A patients at the center were enrolled. all were residents of Duhok.
The patients' demographic information was then collected along with a thorough history and examination of their bleeding tendency (they were asked about their age, gender, age at diagnosis, first bleeding site, frequency of bleeding, family history of diseases with bleeding tendency, first exposure to artificial factor VIII, and treatment they had received), and it was recorded on a special questionnaire form made just for this study.
Then, about five milliliters of peripheral venous blood were taken by clean venipuncture from each patient, 3 mls into a tube containing a Trisodium Citrate anticoagulant, and 2 mls into an EDTA tube, frozen for molecular study. Citrated blood was centrifuged within one hour at 3000 rpm for 20 minutes, and then the plasma was used for immediate performance of PT, APTT, correction study, and factor assay accordingly (An APTT-based assay was used to measure factor VIII level).
The molecular study started with DNA extraction using a modified salt-out method adopted by Iranpur and Esmailizadeh (2010) that yields a very high-purity DNA product [10, 11]. As described previously, the inverse shifting PCR technique was used to detect Inv22 and Inv1 inversion [12]. BclI first digests a genomic DNA sample. This is followed by the self-end ligation of BclI fragments. While irrelevant ligation products are produced as background, some ligation events generate relevant circles that contain the standard FVIII intron 22 and intron 1 sequence or the Inv22 and Inv1 chimers resulting from the intrachromosomal recombination between FVIII intron 22 or intron 1 and its homologous copies outside the FVIII gene. PCR primers (Table 1) [12] are designed to target the BclI spanning regions of these relevant circles. The targeted region of the "normal circle" is amplified by a pair of intragenic upstream and downstream primers. In contrast, that of the "Inv22 circle" and "Inv1 circle" is amplified by an intragenic upstream and an extragenic downstream primer.
The two PCR products are of different sizes, which are resolved by gel electrophoresis. The diagnostic test for Inv22 applies primer ID with a set of three primers U (IU, 2U, and 3U), enabling discrimination of normal allele associated with a signal of 487 bp, from Inv22 type I with a signal of 333 bp and Inv22 type II with a signal of 385 bp. In addition, the diagnostic test for Inv1 applies primer 1 -IU with a set of two primers D (1-ID, 1-ED), enabling discrimination of normal allele with a signal of 304 bp, and Inv1 with a signal of 224 bp. The sample does not prove to be Inv. 1 or 22; send it for sequencing.
Statistical and data analyses were performed using the Statistical Package for the Social Sciences software, version 22 (SPSS Inc., Chicago, IL, USA). The results were reported as mean values ± SD. The t-test and Pearson correlation were used as appropriate, and P < 0.05 was considered statistically significant.
Table 1. The Sequences of Oligonucleotide Primers of Inv1 and Inv22 mutations
|
Mutation |
Primer |
Sequence 5' to 3' |
|
Inv. 1 |
1-IU |
GCCGATTGCTTATTTATATC |
|
1-ID |
TCTGCAACTGGTACTCATC |
|
|
1-ED |
GCCTTTACAATCCAACACT |
|
|
Inv. 22 |
ID |
ACATACGGTTTAGTCACAAGT |
|
IU |
CCTTTCAACTCCATCTCCAT |
|
|
2U |
ACGTGTCTTTTGGAGAAGTC |
|
|
3U |
CTCACATTGTGTTCTTGTAGTC |
Results and Discussion
Eighty hemophiliac patients registered at the Jin Center for Hematological Disease in Duhok/Iraq were included in the current study, ranging in age from 1.4 to 58 years with a median age of 15 years (Mean 17.1±12.2 SD). The majority were in the second decade of their life, and all were males. Their ages at diagnosis vary from 1 month to 18 years, with a median age at diagnosis of 6 months (1.5 ± 0.35 years), and only 22.5% of patients (18 patients) did not have a prior family history (new mutation).
The chief clinical manifestations included a tendency to bleed spontaneously into the skin and mucous membranes, followed by bleeding after slight trauma (Table 2). The presenting symptom was rarely severe bleeding in the form of hemarthrosis, cerebral hemorrhage, or deep muscle hematoma. All patients got factor VIII concentrate on demand, and no inhibitor was found among the enrolled patients (Table 1). The most common chronic sequelae among the recruited patients were joint complication (targeted joint), which was seen in 56 individuals (70%) and predominantly involved the knee, elbow, and ankle joints. Involvement of several joints seen in 11 individuals. Chronic pain was documented in 20 patients (25.0 %), while 60 patients (75.0 %) had no chronic pain. Mostly observed in patients with severe hemophilia, particularly those with multiple joint involvements (p-value 0.0012). Other complication includes viral infection, particularly HBV and HCV infection. Viral hepatitis was only observed among adult patients aged > 16. Of these patients, 8 patients (10%) had viral hepatitis. No patient had an HIV infection. The clinical state of the majority of patients was moderate, followed by severe, and the least frequent was mild (Table 3).
Table 2. Initial clinical presentation that brings a child into diagnosis.
|
First clinical presentation |
|
||
|
Skin |
Ecchymosis |
44 |
45 (56.25%) |
|
Nail bleeding |
1 |
||
|
Following minor trauma or surgical intervention |
Circumcision |
19 |
21 (26.25%) |
|
Trauma to Rt. Ear |
1 |
||
|
Tooth Extraction |
1 |
||
|
Mucus membrane |
Mouth injury |
5 |
7 (8.75%) |
|
Epistaxis |
1 |
||
|
Anal fissure bleed |
1 |
||
|
Joint |
Hemarthrosis |
3 |
3 (3.75%) |
|
ICH and cephalhematoma |
ICH |
2 |
3 (3.75%) |
|
Cephalhematoma |
1 |
||
|
Muscle |
D Muscle hematoma |
1 |
1 (1.25%) |
Table 3. Main Clinical Characteristics of all patients.
|
Parameters |
Severe |
Moderate |
Mild |
ANOVA P value |
|
|
No. (%) |
31 (38.75) |
35 (43.75) |
14 (17.5) |
|
|
|
Factor level (mean%±SD) |
0.61±0.04 |
2.04±0.15 |
13.11±4.05 |
0.002 |
|
|
Age |
26.9±3.4 |
22.3±5.3 |
30.1±4.1 |
0.216 |
|
|
Age at diagnosis |
11.1 ± 3.1 |
14.2 ± 3.8 |
18.1 ±3.8 |
0.034 |
|
|
Chronic pain No. (%) |
12 (38.7) |
7 (20.0) |
1 (7.1) |
0.024 |
|
|
Joint Impairment |
14 (45.2) |
12 (34.3) |
2 (14.2) |
0.0378 |
|
|
Target joint |
26 (83.9) |
26 (74.3) |
5(35.7) |
0.0211 |
|
|
Poor socioeconomic state |
12(38.7) |
15 (42.9) |
4 (28.6) |
0.769 |
|
|
Bleeding in last 4 weeks |
1.83±0.27 |
0.98±0.17 |
0.22±0.78 |
0.013 |
|
|
Viral Hepatitis |
B |
1(3.2) |
1(2.9) |
0 |
Small no. |
|
C |
2 (6.5) |
1(2.9) |
1(7.1) |
||
|
B+C |
0 |
2 (5.7) |
0 |
||
|
Mode of therapy |
On-Demand |
28 |
33 |
14 |
Small no. |
|
Prophylaxis |
3 |
2 |
0 |
||
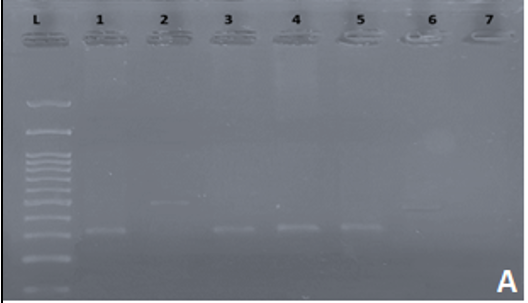
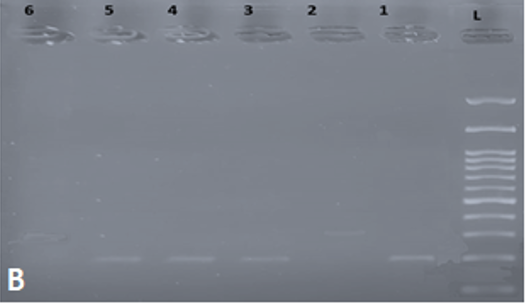
Following PCR and gel electrophoresis, hemophilia patients with Inv22 produce a single band of either 333 bp (Type I) or 385 bp (Type II), while normal individuals produce a single band of 487 bp (Figure 1a). Cases positive for Inv1 produced a single band of 224 bp, while cases that were negative for Inv1 produced a single band of 304 bp (Figure 1b). Following inverse shifting PCR, 35 cases (43.75%) turned out to be inversion 1, 32 cases (40%) turned out to be inversion 22, and the remaining 13 cases (16.25%) remained uncharacterized. The uncharacterized cases were then sent to special laboratories in Korea for sequencing. Some examples of sequencing analysis are seen in Table 4. One single point mutation detected in exon 14 that was not previously reported may be a new mutation specific to the region (Table 4).
Table 4. shows an example of a point mutation in exons 10, 13, and 14 that was associated with mild hemophilia.
|
Sample No. |
Exon |
SPNs |
Mutation |
|
17 |
No specific mutation detected |
||
|
24 |
10 |
rs1428927109 |
G>A |
|
27 |
14b |
COSV64270860 |
G>A |
|
33 |
14c |
Rs1557278336 |
A>G |
|
14b |
COSV64270860 |
G>A |
|
|
34 |
10 |
rs782407122 |
G>A |
|
14b |
COSV64270860 |
G>A |
|
|
36
|
10 |
rs782407122 |
G>A |
|
14b |
COSV64270860 |
G>A |
|
|
38 |
14b |
COSV64270860 |
G>A |
|
61 |
14A |
Rs1557278398 |
G>A |
|
14b |
COSV64270860 |
G>A |
|
|
13 |
rs782026563 |
T>C |
|
|
68 |
10 |
rs782407122 |
G>A |
|
69 |
No specific mutation detected |
||
|
72 |
14b |
COSV64270860 |
GA |
|
74 |
No specific mutation detected |
||
|
|
|
|
a) |
b) |
|
Figure 1. gel electrophoresis following inverse shifting for Inv 22. And Inv1. a) Inv. 22: L: 100-1500 bp ladder; Lane 1: positive control; Lane 2: normal control (Negative for Inv 22); Lane 3-5: positive samples for Inv 22; Lane 6: Negative for Inv 22. b) Inv 1. L: 100-1500 bp ladder; Lane 1: positive control; Lane 2: normal control (Negative for Inv 1); Lane 3-5: positive samples for Inv 1; Lane 6: Negative for Inv 1. |
|
Using the ANOVA analysis test for several samples, Inversion 1 and 22 were associated significantly with severe hemophilia compared to point mutations associated with mild hemophilia with a P value consistently less than 0.05. Using ANOVA study for several samples and many tests, including the Mann-Whitney pairwise test, Kruskal Wallis test for equal medians, and Dunn's post hoc, all show a significant correlation between the type of mutation to the residual Factor VIII level and severity of hemophilia.
The correlation of specific gene mutation with symptoms, signs, and complications is seen in Table 5. Using an ANOVA study for several samples reveals that the type of mutation significantly impacts the frequency of bleeding in the last 4 weeks, development of targeted joints, chronic pain, and development or acquisition of viral hepatitis. Types of the underlying mutations did not impact the demographic data, including age, age at presentation, religion, socioeconomic state, family history, and initial clinical manifestation.
Table 5. Correlation of specific gene mutation with symptoms, signs, and complication
|
Parameters |
Intron 1 |
Intron 22 |
Others |
P. value |
|
Age |
2.3-58 (14.6±14.7) |
2.2-47 (14.9±9.8) |
1.4-37 (17.4±9.74) |
0.345 |
|
Age at diagnosis |
0.01-18 (2.5±4.47) |
0.01-6 (0.7±1.084) |
0.01-5 (0.99 ± 1.46) |
0.567 |
|
Religion (M/Y/M) |
25/8/2 |
24/6/2 |
9/4/0 |
0.453 |
|
Positive Family hx |
26/35 |
23/32 |
13/13 |
0.223 |
|
Positive consang. marriage |
18/35 |
17/32 |
2/13 |
0.124 |
|
Initial presentation |
||||
|
Skin |
22 |
17 |
6 |
0.224 |
|
Following minor trauma or surgery |
10 |
8 |
3 |
0.334 |
|
Mucus membrane |
3 |
2 |
2 |
0.578 |
|
Joint |
2/35 |
1/32 |
0/13 |
Small no. |
|
ICH and cephalhem. |
1 |
2 |
0/13 |
Small no. |
|
Muscle |
0 |
1 |
0 |
|
|
Targeted joint |
||||
|
Absent |
9 |
10 |
4 |
0.023 |
|
Ankle |
2 |
1 |
1 |
|
|
Elbow |
5 |
3 |
0 |
|
|
Knee |
14 |
17 |
3 |
|
|
Multiple Joints |
5 |
6 |
0 |
|
|
Viral Hepatitis |
|
|
|
|
|
Hepatitis B |
0 |
2 |
0 |
0.042 |
|
Hepatitis C |
1 |
2 |
1 |
|
|
Hepatitis B+C |
1 |
1 |
0 |
|
|
Chronic pain |
6 |
12 |
2 |
0.017 |
|
Factor level (%) |
0.1-20 (3.06±4.42) |
0.2-11.7 (3.46±2.97) |
0.1-50 (6.55±13.95) |
|
|
Poor social state |
14/35 |
13/32 |
4/13 |
|
|
Bleeding, last 4 weeks |
1.97±0.19 |
1.67±0.54 |
0.13±0.12 |
0.022 |
|
Mode of therapy Prophylaxis |
3/35 |
2/32 |
0/13 |
Small no. |
According to the World Federation of Hemophilia (WFH), around 400,000 people have hemophilia, with just 25% receiving adequate treatment. Most children with severe hemophilia would die young if not treated; however, if appropriately treated, people can live normal, healthy lives [13].
The clinical features at diagnosis vary depending on the community. Apart from a positive family history, the most common presenting signs in the current study were modest bleeding manifestations. This conclusion is congruent with a study from Iraq/Sulaymaniyah [6], which showed that most patients presented with cutaneous symptoms, followed by mild trauma such as circumcision. They reported that 25% of patients presented with joint swelling when they first arrived. Our finding was also in concordance with that reported from Germany, in which the reason for diagnosis in the majority of the cases 47% were hematomas, followed by family history 24%, operations 14%, joint bleeding, muscle bleeding, and mouth bleeding each accounted for 5% [14]. The variation in the presenting features may reflect the prior family history, parents' educational attainment, access to health services, severity of the disease, and the underlying mutations. Major and serious bleeding like muscle hematoma and intracranial bleeding constitute the minority of presenting features worldwide, including in Egypt and India [14-16].
According to the Universal Data Collection (UDC) from the USA, patients with severe hemophilia had a higher risk of developing a target joint than those with moderate or mild hemophilia (33.1% versus 18.8% and 5%) [17]. According to the WFH 2005 report and following the current study, the joints that are most frequently afflicted in individuals who are not receiving treatment are the knees (45%), elbows (30%), ankles (15%), shoulders (3%), and wrists (2%). The same is reported by Islam et al. 2022 from Bangladesh and by Mohsin et al. 2010 from Pakistan [18, 19].
A major side effect of replacement therapy is the development of FVIII inhibitor, which affects 20–35% of patients with severe HA and 3–13% of those with mild-to-moderate HA [20, 21]. All participants in the current study had an APTT-based test for coagulation inhibitors, which failed to detect inhibitors in all recruited patients, and all had negative family histories for inhibitors despite the expectation that there would be inhibitors, particularly in severe cases. This may be attributed to a number of factors, including the reduced usage of replacement therapy [5, 22].
The higher rate of patients with moderate disease in our locality can be due to the type of causative FVIII mutations or to some unidentified modifier agents among people in the region. Moreover, the discrepancy in FVIII activity as measured by one-stage and two-stage assay could be the underlying reason for the high percentage of moderate disease [23, 24].
Viral hepatitis is considered one of the expected complications of hemophilia, especially among older patients who previously received locally made products from human sources. In Sulaymaniyah, 6 out of 126 cases of hepatitis were found, of which 5 were hepatitis B (4%) and 1 was hepatitis C 1(0.8%). From 229 cases of hemophilia A patients in Pakistan, hepatitis B virus was found in 7 severe cases and 3 moderate cases, while hepatitis C virus was found in 59 severe (65.55%), 24 moderate (26.66%), and 7 mild cases (7.77%) from total 90 persons infected. HIV was detected in two patients with a severe form of hemophilia, and no case was found to have a combined infection [6, 19].
Surveys in Europe by Hohlstein on chronic pain in connection with hemophilia show that 32–50% of hemophilia patients suffer from arthropathy-related pain, which leads to physical inactivity and therefore also an increased risk of cardiovascular events and has a negative influence on psychological health and the survey included 1678 children (47% severe hemophilia, 8% with chronic pain and 5103 adults (44% severe hemophilia, 35% with chronic pain) [25]. Also, a patient survey in Germany by Kalnins illustrated how pain affects the patient's quality of life; of the patients surveyed, 86% suffered at least occasionally from hemophilia-related pain [26]. A previous study in Iraq/Duhok shows a significantly lower score of health-related quality of life in hemophilic patients in regards to lower physical activity, emotional changes, and general health, especially from a positive family history. Chronic pain was the reason for limiting physical activities and emotional changes [5].
In our study, a limited number of patients are on prophylactic treatment, which includes 3 of the severe cases and 2 of moderate severity, while the majority are on the demanded type of therapy as reaching access to the continuous type is inadequate or not available and expressive. In a previous study, Iraq shows that the number of patients on continuous (prophylaxis) 45 (35.7%), while on episodic (On demand) 81 (64.3%), and complications mostly involve the on-demand type of therapy. In contrast, in developing countries, patients in developed countries have much better access and more resources to provide continuous types of therapy for their patients and, hence, reducing the lifelong complication of hemophilia [6].
Molecular genetic testing is performed on probands to detect the family-specific mutation in FVIII. Identification of the specific FVIII mutation can help predict the clinical phenotype and assess the risk of developing an FVIII inhibitor and can even possibly be used to predict responsiveness to immune tolerance induction. Carrier testing for at-risk relatives and prenatal diagnosis for at-risk pregnancies requires prior identification of the disease-causing mutations in the family if the direct mutation detection strategy is used [4].
In a study conducted in Pakistan, 92 HA patients were screened for the F8 gene mutation. Mutations in the F8 gene are heterogeneous and include point mutations (including missense, nonsense, and splice site mutations), inversions (Inv22 and Inv1), and frameshifts (deletions and duplications). 42% of HA patients had point mutations, 20% had Inv22, and 29% had severe disease [27].
In our study, the results show Inv1 mutation detected in (35/80 patients) 43.75%; 17 cases had a severe form of HA (48.57%), 12 moderate cases (34.28%), and 6 cases of mild form 17.1%. The mutation is seen less commonly in other populations; from a Jordanian study of 142 HA patients, 117 cases of severe HA, and 2% of severe HA patients have Inv1. In addition, they also reported 19 different mutations, including point mutations and frameshift mutations, including 15 new mutations [28]. In Lebanon, the authors found that 23 out of 79 patients (29%) had Inv.22, and 2.5% had Inv1mutatin. In addition, the authors reported 32 different mutations comprising single point mutations, small and large deletions, and splice mutations. Of the 32 mutations, 21 mutations were novel [29]. From Saudi Arabia, Inversion 22 mutation was found in 15 out of 110 patients (13.6), 2 patients (1.8 %) showed Inv1, and out of 32 cases sequenced for coding exons, 2 novel mutations were found [30]. The Tunisian study reported 23 different FVIII gene mutations in 28 HA patients. The identified mutations included 5 Inv 22, 7 insertions, 4 deletions, and 7 substitutions. The distribution of mutations (n = 18) beyond inv22 showed that 9 were located in exon 14, the most mutated exonic sequence of the F8 gene, and 8 were novel mutations [31]. A study from France analyzed 120 HAs from 94 unrelated families and identified 47 mutations in the F8 gene, including 26 novel mutations. Inv22 was detected in 47% of patients, and no patients in this study had inv1 [32].
These and other studies' results further emphasize HA's genetic heterogeneity. More than 1200 mutations in the F8 gene are listed in the HAMSTeRS database. Among these, the most common defect is an inversion in intron 22 and intron 1. The intron 22 inversion is detected in 40–50% of severe HA patients. However, the percentage varies among different populations. Differences in reported prevalence rates from different countries were attributed in some studies to the limited number of studied cases, the ethnic variations, and the inclusion of patients with moderate and mild FVIII activity results [33].
Conclusion
Molecular heterogeneity of the underlying mutations in hemophilia A in various populations and the mode of therapy contributes to the variation in the presenting clinical features of short-term and long-term complications in affected patients.
Acknowledgments: None
Conflict of interest: None
Financial support: None
Ethics statement: The current study was approved by the ethical committee at the Directorate of Health/Duhok and the scientific committee of the College of Medicine/University of Duhok. At first, all patients were informed about the study; written consent was obtained from all patients.