Pharmacophore an International Research Journal
CORPUS CALLOSUM AGENESIS WITH CHORIORETINAL ABNORMALITY (AICARDI SYNDROME): AN EDUCATIONAL REVIEW
Lalrameng Mawii, Khayati Moudgil*
|
|
Research Scholar, Department of Pharmacology, JSS College of Pharmacy, Ooty, India. JSS Academy of Higher Education and Research. |
ABSTRACT
According to the Office of Rare Diseases (ORD) of the National Institutes of Health (NIH), Aicardi syndrome is a genetic condition affecting about 1 in 105,000 to 167,000 new-borns in the United States. This disease primarily affects females and is characterized by infantile spasms, partial or complete agenesis of corpus callosum (white holes) and characteristic lucan-shaped chorioretinal lesions (eye defects). Signs normally occur between the ages of 2 and 5 months, marked by a form of seizure in children, wheezing, and the appearance of yellowish spots in the eye, which may lead to epilepsy in later stages. There is no particular medication available for Aicardi Syndrome, but signs and causes can be resolved through therapy.
Keywords: Aicardi Syndrome, Infantile spasms, Chromosomes, Female
Introduction
Aicardi syndrome is a severe neurodevelopmental condition that interferes with the formation of the corpus callosum and was first described by the French neurologist Dr. Jean Aicardi (1926-2015) in 1965 [1]. This is associated with a lack of tissue that binds the left and right half of the brain (agenesis of the corpus callosum), infantile spasms (seizure) that can lead to epilepsy (recurrent seizures) [2], chorioretinal lacunae (defects of light-sensitive tissue in the retina) as seen in Figure 1, eye anomalies with poorly formed eyes, and optic nerve gaps that can also lead to blindness. Severity and symptoms vary where some people die during childhood and few survive until they become adults. Aicardi syndrome is severely associated with high mortality, poor prognosis, and developmental outcomes.
Etiology of Aicardi syndrome (why females have a high risk of infection?)
The main cause is unknown and mainly affects females, believed to be due to mutation in the X chromosome gene. An average human has 46 chromosomes, with males having XY and females having XX. There is inactivation of one of the X chromosomes in Aicardi syndrome, so each female (XX) and male (XY) have only one functioning X chromosome. X-inactivation happens mostly at random, but in a few cases, it does not occur at random and is known as distorted X-inactivation. Skewed X-inactivations are primarily found in girls with significant gene mutations in each of the X chromosomes. Most cases are not inherited as they are caused by new gene mutations and can occur in a family with no history of disorder. Aicardi syndrome also affects male babies who have an extra copy of XXY X chromosome and are also known as Klinefelter syndrome [3].
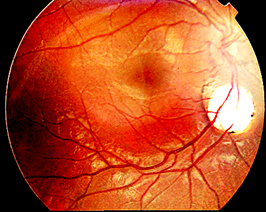
Figure 1: Chorioretinal lacunae [4]
Prevalence and incidence of Aicardi syndrome
The exact incidence and prevalence are uncertain. Lund et al. performed an epidemiological study of females (0-29 years of age) in Norway in 2011, with a prevalence rate of 0.63 per 100,000 females [5]. A study was performed in 2008 to determine the incidence and prevalence of Aicardi syndrome in the age group from 1 month to 42 years of age. Incidence was estimated to be 1 in 105,000 in the United States and 1 in 93,000 in the Netherlands. The chances of survival of patients at 27 years of age were found to be 62 percent [6].
Diagnostic criteria
Imaging studies-Neuroimaging, MRI, CT-Scan and Simple Radiographs help to determine autism, developmental delay, agenesis of the corpus callosum, cortical migration defects, congenital infections, and skeletal malformations. Other checks include ophthalmological evaluation, electroencephalography (EEG) [7-10].
How to manage and what should be the treatment goals?
There is currently no known treatment for Aicardi Syndrome, but symptoms can be controlled depending on the conditions of the patient. Patients with seizures may be treated with antiepileptic medications or with a ketogenic diet. Physical, occupational, and speech therapy can be useful for recovery. Gastrointestinal conditions can be treated by a gastroenterologist; palliative epilepsy surgery and nerve stimulation are also effective in some cases.
Pharmacological management [11]:
a. Corticotropin (Corticosteroid):-
b. Vigabatrin (Anticonvulsant):-
DISCUSSION AND CONCLUSION
Few case reports have been reviewed and the information collected is shown in Table 1. The studies published in the literature all suggest that females are more likely to suffer from Aicardi syndrome.
Table 1: Case reports collected on Aicardi syndrome.
|
Author |
Jean Carlos de Oliveira Menezes et al. [11] |
Parag K Shah et al. [12] |
Jennifer Dennis B. D. Bower [2] |
|
Year |
2018 |
2009 |
1972 |
|
Chromosome |
46 XX |
46 XX |
46 XX |
|
Sex |
Female |
Female |
Female |
|
Age |
2 months |
3 months |
2 days old |
|
Gestation (weeks) |
40 weeks (Caesarean) |
40 weeks (Normal) |
40 weeks (Spontaneous delivery) |
|
Birth weight (g) |
3.760 |
- |
3.920 |
|
Height |
50 cm |
- |
- |
|
Age of onset |
2 months |
1 and a half months |
Second day of life |
|
Symptom |
Delayed neuropsychomotor, infantile spasm, supine posture, absence of spontaneous movement, presence of a vesicular murmur |
History of flexor spasm, seizure, twitching of both eye 5-6 times, drooling of saliva, apparent blindness |
Twitching of eyelid and left eye, mild seizure, tonic seizure at 3 weeks, infantile spasms began at 7 weeks, the head is misshapen, facial asymmetry, flattening of the left side of the face |
|
MRI |
Corpus callosum agenesis |
- |
- |
|
Management |
Nutrition, Psychologist, Speech therapist, Exercise, Phenobarbital, Baclofen (spasticity) |
Anti-epileptic, Subcutaneous injection of adrenocorticotropic hormone (ACTH) |
Nitrazepam, ACTH gel 10 units two times a day for 5 days, Prednisolone 2.5mg two times a day till 4 months |
|
Examination |
Eye Movement Exam (EME), Biomicroscopy exam (BCE) - transparent cornea & crystalline, Fundoscopy Exam (FE) - retina with normal staining, absence of chorioretinal lacunae. |
Development delay, head circumference-10th percentile Ophthalmic examination- not respond to bright light, pupils were sluggish Fundus examination- Bilateral optic nerve colobomas, presence of chorioretinal lacunae EEG- Burst of spike, polyspike, sharp & slow-wave complexes with suppression, Bilateral synchronous & symmetrical discharge CT Scan- Hypogenesis of the corpus callosum with a small inter-hemispheric cyst. |
Ophthalmic examination- The optic disc was funnel-shaped and dark. Scoliosis of the dorsal spine, convex to the right. Hb-14.0 g, WBC-9,000/mm3, Blood plasma calcium-5.1mEq 1 litre, Random blood sugar-95 mg/100 ml, Protein-40mg/100ml. X-ray- Multiple congenital defects of mid-dorsal vertebrae, spine bifida, abnormal ribs. During encephalopathy cardiac arrest occurred. EEG- Presence of hypsarrhythmia-chaos and high amplitude |
There are few organizations and groups, which support the patients who suffer from Aicardi syndrome, which are presented in Table 2.
Table 2: Organizations and groups supporting Aicardi syndrome that can help to connect with other patients and families.
|
1 |
Aicardi Syndrome Foundation P.O. Box 3202 St. Charles, IL 60714 Telephone: +1-630-397-9728 |
|
2 |
National Organization of Disorders of the Corpus Callosum PMB 363, 18032-C Lemon Drive Yorba Linda, California 92886 Telephone: (714) 747-0063 Fax: +1-714-693-0808 |
The review article shows the relevance and management of Aicardi Syndrome and offers understanding within the medical community.
Conflict of interest
The authors declare none.
References


